Spinalna mišićna atrofija (SMA) rijetka je, nasljedna, autosomna recesivna neuromišićna bolest koju karakterizira degeneracija alfa motoričkih neurona u prednjim rogovima kralježnične moždine, koja dovodi do klinički progresivne atrofije mišića, mišićne slabosti i gubitka pokretljivosti. Najčešći je oblik te bolesti SMA povezan s mutacijama na kromosomu 5q, koji čini približno 95% slučajeva.1,2
SMA je danas jedna od najčešćih i najozbiljnijih genskih bolesti u dječjoj dobi. Za tu je bolest karakteristično da djeca imaju izrazitu slabost gornjih i donjih ekstremiteta, a moguće i slabe bulbarne i dišne mišiće, dok kognitivna sposobnost tih bolesnika naizgled nije narušena.2–6
Trenutno se bolesnici oboljeli od SMA razvrstavaju u pet kliničkih potkategorija (tip 0, I, II, III i IV), definiranih prema dobi pri nastupu simptoma bolesti, težini bolesti i najvećem stupnju postignutih motoričkih sposobnosti. Kod SMA tipa 0 klinički su znakovi primjetni već pri rođenju i to je najteži oblik bolesti, kod koje je očekivani životni vijek kraći od 6 mjeseci.2,7
Procjenjuje se da je globalna prevalencija SMA približno 2 – 6 na 100 000 ljudi, dok procijenjena globalna incidencija na 100 000 živorođene djece iznosi 3,5 - 7,1 za tip I, 1,0 - 5,3 za tip II te 1,5 - 4,6 za tip III.2,7,8 U Europi se godišnja incidencija razlikuje od države do države, kao i prema fenotipu.9
Genska komponenta:
Godine 1995. ustanovljeno je da SMA uzrokuje homozigotna mutacija i/ili delecija gena na kromosomu 5q13, danas poznatom pod nazivom gen za preživljenje motoričkih neurona-1 (SMN1). Ta je genska promjena odgovorna za nedovoljnu proizvodnju SMN proteina, koji je ključan za održavanje motoričkih neurona, što dovodi do progresivne degeneracije tih neurona u kralježničnoj moždini.4,10,12
Međutim, prisutnost drugog gena SMN (SMN2), koji je gotovo identičan genu SMN1 jer se od njega razlikuje u samo 5 nukleotida, omogućuje stvaranje manjih količina SMN proteina. Pritom se proizvodi oko 90% nestabilnog SMN proteina, koji nije funkcionalan i podliježe brzoj razgradnji, te 10% normalnog SMN proteina.2,7,8,10,12
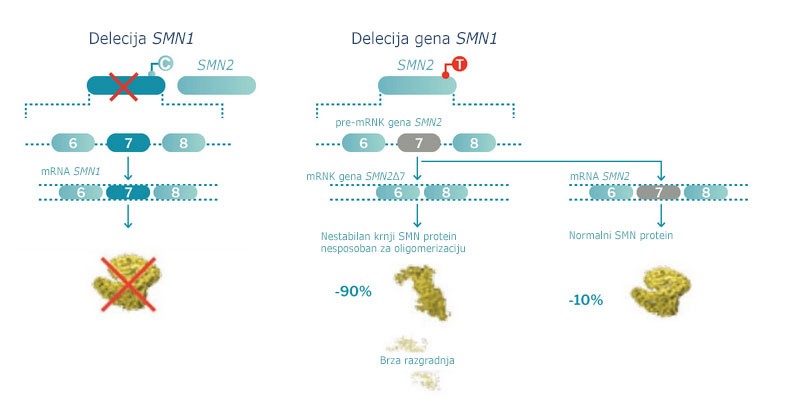
Slika 1. Normalna proizvodnja SMN proteina translatiranog iz gena SMN1 u usporedbi s alternativnim putem, odnosno proizvodnjom istog tog proteina translatiranog iz gena SMN2. Bolesnici oboljeli od SMA nemaju nijedan gen SMN1 i oslanjaju se samo na alternativni put proizvodnje SMN proteina. pre-mRNK: predglasnička ribonukleinska kiselina; mRNK: glasnička ribonukleinska kiselina
S tim u vezi treba naglasiti da je broj kopija gena SMN2 u korelaciji s dobrom prognozom i fenotipom SMA, čak i ako je proizvodnja funkcionalnog proteina iz tog gena nedostatna za preživljenje neurona u središnjem živčanom sustavu (SŽS).1,12